
25
Mar
Introducción al RNA-seq
¿Que es el RNA-seq?
El RNA-seq es una técnica de análisis del transcriptoma. Aunque a día de hoy existen muchas variantes de la misma, en general se puede describir como la secuenciación de ARN mediante tecnología de secuenciación masiva, las conocidas NGS, de tal forma que se obtenga información acerca de la secuencia y cantidad de estos transcritos. En base a estos dos datos podremos determinar el grado de expresión de cada transcrito y, si además comparamos dos condiciones, es posible determinar las diferencias entre los distintos perfiles de expresión para dichas condiciones.
Esta es una técnica revolucionaria ya que permite observar fenómenos de splicing, modificaciones post-transcripcionales, fusión génica, mutaciones/SNP y cambios en la expresión génica a lo largo del tiempo, todo ello en un solo experimento.
Esta gran cantidad de información conlleva a que el flujo de trabajo sea amplio y complejo. Desde el diseño experimental y la extracción de ARN, hasta la lista de genes diferencialmente expresados, hay que pasar por multitud de etapas tanto de laboratorio como de computación. Debido al enorme crecimiento de este tipo de análisis, también han crecido las técnicas computacionales asociadas, dando lugar a la bioinformática tal y como la conocemos hoy.
La bioinformática asociada a las tecnologías NGS es por tanto una disciplina joven, y carente de estandarización a la hora de abordar un problema. Es por ello que es común que distintos investigadores programen cada uno su propia solución a una misma cuestión biológica. Siendo en la mayoría de casos, igual de válidas las distintas herramientas desarrolladas.
Esto supone un problema para posteriores investigadores que se ven en la tesitura de no saber que herramienta elegir a la hora de llevar a cabo sus análisis. Para más inri, la respuesta a esa pregunta suele ser: “la que mejor resultados proporcione”. Y es que, como es de suponer, aunque dos softwares estén diseñados para una misma tarea, sus resultados nunca serán idénticos.
Y es, precisamente, en estos dos aspectos, tanto para llevar a cabo la mejor estrategia de secuenciación, como para poner en marcha el análisis de datos más acorde, donde es más que recomendable contar con el asesoramiento y la ayuda de especialistas.
Pasos de un RNA-seq
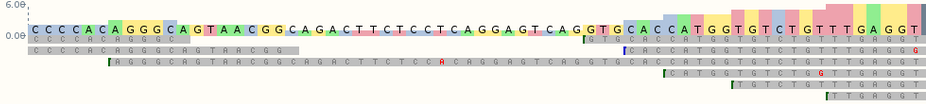
En todos los casos, un experimento de RNA-seq implica la creación de una colección de fragmentos de ADNc que están flanqueados por secuencias específicas (conocidas como adaptadores) que son necesarias para la secuenciación. Este conjunto (denominado biblioteca) se secuencia por lectura corta, produciendo millones de lecturas de secuencias cortas que corresponden a fragmentos denominados shotgun.
Un análisis típico de ARN-seq cuenta con los siguientes pasos

Cada uno de estos pasos tiene multitud de variantes, de entre las cuales el investigador ha de decidir con el objetivo de responder de la manera más específica posible a las preguntas de investigación que plantee. De lo contrario, puede suponer un importante gasto en tiempo y recursos. Así pues, vamos a hablar de algunos de los pasos que más interés puedan suscitar a las personas que se estén planteando llevar a cabo un RNA-seq.
Identificación del objetivo del estudio
Mediante la técnica RNA-seq se puede obtener dos tipos de información, cualitativa y cuantitativa
Cualitativa. Los datos incluyen la identificación de los transcritos expresados, y la identificación de los límites exón/intrón, los sitios de inicio transcripcional (TSS) y los sitios de poli-A. Nos referiremos a este tipo de información como «anotación».
Cuantitativa. Los datos incluyen la medición de las diferencias de expresión, splicing, TSS alternativo y la poliadenilación alternativa entre dos o más tratamientos o grupos. Aquí nos centramos específicamente en experimentos para medir la expresión génica diferencial (DGE).
Estas dos propiedades funcionan como las dos caras de una misma moneda. Por ejemplo, una anotación de calidad necesita cierto grado de cuantificación, el hecho de que un par de lecturas mapeen en una zona intergénica no es suficiente para considerar la existencia de un gen.
A la hora de diseñar el análisis, el investigador deberá decidir en qué grado quiere desarrollar cada uno de los posibles tipos de información.
Enriquecimiento específico.
Es necesario procesar el ARN extraído total, ya que normalmente cerca de más del 80% de la muestra será ARNr. Algunos de los métodos de eliminación de ARNr son los siguientes.
Hibridación vía oligo-dT. Mediante secuencias complementarias a la cola poli-A característica de los ARNm eucarióticos. Es esencial tener en cuenta el organismo que estamos analizando, así como todos los transcritos que carecen de cola poli-A
SuperSAGE. Es una aproximación que basa la secuenciación del transcrito en una pequeña zona de su extremo 3’. Tiene un gran potencial, pero necesita que la anotación del genoma esté especialmente completa, ya que suelen flaquear precisamente en las zonas 3’ UTR. Otra desventaja es que se trata de una técnica insensible a splicing.
Eliminación del ARNr por hibridación. Existen kits comerciales basados en secuencias complementarias a zonas altamente conservadas de ARNr que permiten la efectiva eliminación de los mismos. En contraposición a la hibridación vía oligo-dT expuesta más arriba, en este caso no se pierden los transcritos no poliadenilados.
Selección de tamaño. Tradicionalmente la selección de ARN de pequeño tamaño se llevaba a cabo mediante la recuperación de las bandas de interés de electroforesis, pero debido a los inconvenientes que se planteaban, se han desarrollados kits comerciales basados en extracción en fase sólida.
Estrategia de retro-transcripción
Todos los protocolos actuales utilizan la capacidad de la transcriptasa inversa (RT) para sintetizar una cadena de ADN utilizando el ARN como cadena molde. La RT, al igual que otras polimerasas, requiere un cebador para iniciar la polimerización. Se utilizan varias opciones de inserción del cebador para la primera cadena. Estas incluyen las siguientes:
Uso de oligos-dT. Tiene la ventaja que es independiente de la secuencia del transcrito, pero solo es posible para aquellos que contienen la cola poli-A

Uso de oligos aleatorios. Se utilizan cebadores con secuencias aleatorias. Esto, en principio, permite la secuenciación de todos los transcritos, sin embargo, se ha demostrado que es un sistema dependiente hasta cierto punto de la secuencia. Por lo que se sobre-representan ciertos ARN generando hotspots

Uso de oligo pre-ligado. Es la técnica más usada a día de hoy, consiste en ligar un adaptador con una secuencia conocida al extremo 3’ del ARN usando T4-RNA ligasa. Esto tiene la ventaja de reducir el sesgo de cebador, ya que todos los ARN se preparan utilizando la misma secuencia pudiendo retener información específica de a que cadena pertenece cada transcrito

¿Qué tendremos la semana que viene?
Siguiendo con la línea del RNA-seq y la enorme cantidad de decisiones que hay que tomar en torno a esta técnica, hablaremos de uno de los principales pasos de su análisis bioinformático, el mapeo. Además, compararemos dos de las herramientas más utilizadas para ello, tanto para resaltar sus diferencias como para ilustrar en qué punto se encuentra la bioinformática a día de hoy.
¡Os esperamos!