23
Jun
Variant Calling
¿Qué es el Variant Calling?
Se define como el proceso mediante el cual se identifican variantes a partir de datos de secuenciación. El proceso que se lleva a cabo groso modo de la siguiente forma:
- Obtener datos en formatos FASTQ de los datos de secuenciación de genoma o exoma completo.
- Alineamiento de las secuencias a un genoma de referencia por medio de archivos formato BAM o CRAM.
- Identificar donde las lecturas alineadas se diferencian del genoma de referencia y guardar el resultado en un archivo formato VCF.
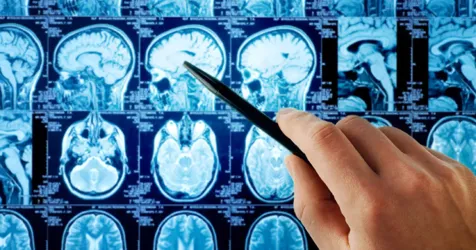
Las variantes se representan en el llamado formato VCF (Variant Call Format). La obtención de este archivo suele obtenerse por el uso de un “variant caller” también conocida como “SNP caller” a partir de uno o más archivos de alineamiento BAM o CRAM. En la imagen inferior se muestra un archivo CRAM típico obtenido al alinear las lecturas a un genoma de referencia.
¿Por qué es importante el Variant Calling?
Permite la identificación de polimorfismo de un solo nucleótido (SNPs) e inserciones y deleciones (indels) a partir de datos de secuenciación masiva (NGS). Es un análisis esencial en el campo de la genómica comparativa. La importancia de estudiar variaciones genética está en las asociaciones que puedan tener con el riesgo de desarrollar determinadas enfermedades, la eficacia y efectos secundarios de fármacos, información filogenética entre otros muchos aspectos. Es importante recalcar que a pesar de aportar información muy útil, sigue siendo un análisis bioinformático predictivo. No se deberían de abstraer conclusiones sobre la repercusión fenotípica de una determinada variación genética sin datos experimentales. Por ejemplo, se podría encontrar una variación SNP en una secuencia proteica dada. Se podría predecir pérdida de función pero se requieren datos experimentales para afirmar dicha conclusión puesto que no se conoce en qué manera afecta la mutación a la estructura y función.
Se puede clasificar el Variant Calling en somático y germinal. En Variant Calling germinal el genoma de referencia empleado es el “estándar” para una especie dada lo que permite identificar genotipos. En genomas diploides permite determinar si un individuo para un alelo determinado es homocigoto o heterocigoto.
En Variant Calling germinal el genoma de referencia proviene de un tejido relacionado del mismo individuo. En este caso se busca determinar mosaiquismo entre células.
Existen multitud de “variant callers”. Aquí mencionaremos unos cuantos:
- bcftools. Herramienta bioinformática muy flexible que presenta multitud de funciones para llevar a cabo el Variant Calling y manipular los datos VCF.
- FreeBayes. Herramienta bioinformática que permite la detección Bayesiana de variantes genéticas para encontrar pequeños polimorfismo como SNPs, indels, MNPs.
- GATK (The Genome Analysis Toolkit). Herramienta bioinformática estándar especializada en la detección de SNPs e indels a partir de datos de ADN y ARNseq de línea germinal. Se puede emplear también para Variant Calling somático y detección de otras variaciones como CNV (Copy Number Variation) y SV (Structural Variaton).
Interpretar el formato VCF
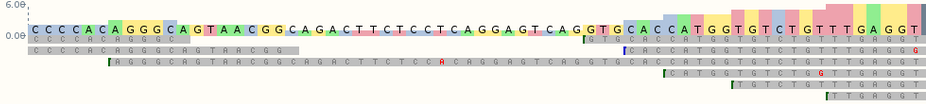
El formato VCF permite de forma sencilla y flexible almacenar las variaciones genómicas. La información se estructura en diferentes columnas como se muestra en la imagen inferior. La explicación de cada uno de los parámetros aparece en la tabla.
![]()
| Tabla del significado de cada columna para el formato VCF. | |
| #CHROM | Cromosoma |
| POS | Posición. Indica donde comienza la variación |
| ID | Identificador |
| REF | Alelo de referencia. Muestra lo que se encuentra en el genoma de referencia. |
| ALT | Alelo alternativo. Muestra lo que se ha encontrado en el genoma estudiado. |
| QUAL | Calidad. Valoración de 1-100 de la calidad. |
| FILTER | Filtro. Indica si ha superado el filtro de calidad |
| INFO | Información. Nos aporta información acerca de la variación |
| FORMAT | Listado (opcional) de descripción de muestras. |
| NA0001 / NA0002 | Identificador individual. Se representa el genotipo de la muestra con un pipeline de tal modo que:
0|0 homocigoto para el alelo de referencia 0|1 heterocigoto 1|1 homocigoto para el alelo alternativo |
Conclusión
En la entrada de hoy hemos visto el WorkFlow general seguido en Variant Calling, entendido su importancia y la estructura del formato utilizado.
¿Qué se va a ver en el próximo blog?
Se verá la automatización del acceso a NCBI por medio de la herramienta Entrez.